My research solves quantitative problems, e.g. electron-phonon coupling and defect thermodynamics, in materials science and physics, from first-principles modelling. The modelling is based on Schrodinger equation involving complex correlation between different particles such as ions, electrons, and phonons. My work provides valuable insights (some has been validated by experiments) into advanced electronic material development.
My research includes:
-
Electronic structures (defect level, band edge position, and band diagram in heterostructures) of semiconductor materials
-
Transport properties i.e. thermal conductivity and electron mobility in electronic materials for electron-phonon scattering
-
Thermodynamics analyses of phase stability and structure-property relationships in materials.
Electronic Structures of semiconductor materials
Materials Challenges in Clean Energy Vision
-
Materials applied in solar cells and power devices play key role in clean energy.
-
Due to the fundamental limit of Si and other conventional semiconductor materials, novel materials are highly desired.
-
Electronic structure, such as band gap, band edge positions, thermodynamics of dopand and defect, etc, are most important feature in determining whether a new material is promising for these applications.
Why First-principles DFT calculations?
Why Schrodinger's equation is unsolvable for normal materials?
Data is too big! For example, wavefunction of two particles in a 1D well can be stored numerically on a 2D mesh. However, for normal materials (e.g. Si which has 84 dimensions), the wavefunction becomes too big to store because the data size increases exponentially with dimension ∝ 10^n.
Density functional theory (DFT) is a computational quantum mechanical modelling method. It can solve the "exponential wall problem" of many-body Schrodinger's equation in correlated systems.
Typical running time on HPC
Using 200 cores: 10 hours using advanced functional for a large system
Storage requirement
10 GB to store a large wavefunction.

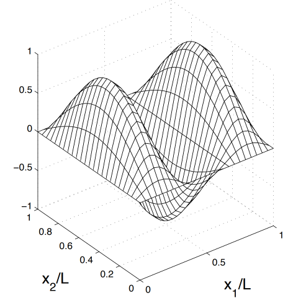
Wavefunction (WF) of two particles in a well calculated using Schrodinger's equation. It can be stored numerically on a 2D mesh.

Comet High-Performance Computing at UCSD
Some Results
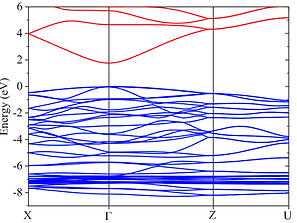
Electronic band structure of ZnSnN2, a promising earth-abundant solar cell materials. The blue lines are valence-bands which are occupied by electrons, and the red lines are unoccupied conduction-band. The gap between the occupied/unoccupied is called band gap.
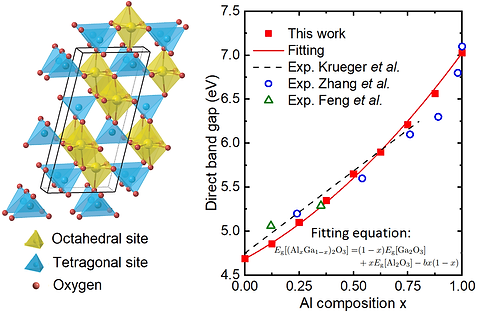
Left: Crystal structure of the ternary alloy (AlxGa1-x)2O3, a promising materials for high-power transisters;
Right: Calculated band gap of the alloys fitted using the bowing equation.
A "non-physical" approach
Data Science and Machine Learning techniques were employed to predict the band gaps of a large number of materials only using their basic information e.g. element and symmetry. Check: Data_Science
Transport Properties of semiconductor materials
Transport properties, i.e. thermal conductivity and electron mobility, are key parameters of semiconductor materials especially for those applied in high-power-high-frequency devices. In addition, the transport coefficients are associated with many advanced properties such as the galvanomagnetic, thermoelectric, and thermomagnetic effects in a semiconductor.
We combined the phonon scattering from EPI and three phonon processes to understand thermal conductivity in electron doped SiC
We also derived phonon-limited electron mobility of ZnO and GaN and discovered that the much lower electron mobility in ZnO attributes to its higher piezoelectric potential and lower longitude optical phonon energy.
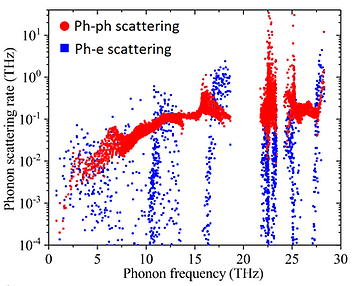
Phonon-electron scattering rates compared to phonon-phonon scattering in SiC